



张路医生的科普号
- 精选 [探秘Castleman病]:初探你可能不太了解的“Castleman病”
百字文摘Castleman病(Castleman Disease,简称CD),又称巨大淋巴结增生症或血管滤泡性淋巴组织增生,是一类罕见的、具有特征性病理改变、高临床异质性的淋巴增生性疾病。目前,Castleman病的诊断和治疗还面临着巨大挑战。近年来 ,Castleman病诊疗领域正在迅速发展,我们对该疾病的了解也越来越多。 研究历史1954年,美国内科医生、病理学家本杰明·卡斯曼(Benjamin Castleman)首次报道并命名了一例局部纵隔淋巴结肿大的单中心Castleman病(UCD)病例; 1969年,Flendrig与Schillings等对Castleman病的组织病理学进行了描述,首次将Castleman病分为浆细胞型、透明血管型和混合型; 1978年,Gaba等首次报道了累及多个淋巴结区域的多中心型 Castleman病(MCD); 1985年,Lachant等首次报道了HIV病人中卡波西肉瘤与MCD的相关性,这一发现启发了随后Soulier等对MCD与HHV-8病毒感染关系的研究,此后抗HHV-8病毒药物被引入Castleman病治疗; 1989年,Yoshizaki首次报道了Castleman病患者中B细胞IL-6表达水平升高,将抗IL-6和IL-6R单克隆抗体引入Castleman病治疗; 近70年的研究与经验积累使得医学界对这一罕见疾病有了更深入的认识:2016年,Castleman病的国际共识定义和诊断标准出台;2017年,针对特发性多中心Castleman病(iMCD)的国际共识诊断标准出台; 2018年,针对iMCD的第一个治疗指南发布。 Castleman病分型Castleman病的临床表现与病理特征多样,对Castleman病的分类/分型是临床诊断和治疗的基础,不同亚型Castleman病在症状,临床表现,疾病机制,治疗方法和预后方面差异很大。 根据淋巴结分布与器官受累部位,Castleman病可分为UCD和MCD。UCD仅累及单个淋巴结区域,全身症状反应较轻,预后较好;MCD累及多个淋巴结区域,多有全身症状,预后较差。 根据病因和临床特点,MCD进一步分为HHV-8相关MCD,POEMS相关MCD和iMCD。其中HHV-8相关MCD常见于HIV感染患者或免疫抑制人群,HHV-8感染被认为是该型MCD的直接病因。POEMS相关MCD是指与POEMS综合征并发的MCD,其临床表现与POEMS综合征重叠,包括多发性周围神经病变(Polyneuropathy) 、器官肿大 (Organomegaly) 、内分泌异常 (Endocrinopathy) 、血清中存在M蛋白 (Monoclonal protein) 和皮肤改变 (Skin changes) 。iMCD则是一类目前病因未明、与POEMS综合征无关的MCD。iMCD又根据临床表现分为TAFRO亚型和NOS亚型。TAFRO亚型的特点为血小板减少 (Thrombocytopenia) 、重度水肿 (Anasarca) 、发热 (Fever) 、骨髓纤维化 (Reticulin fibrosis) 和器官肿大 (Organomegaly) ,其临床表现更重,预后更差;而NOS亚型主要表现为IL-6升高、血小板正常或增多、多克隆免疫球蛋白升高,全身水肿少见(图1) UCD概述UCD是Castleman病最常见亚型,其特征为一个或多个增大的淋巴结仅存在于单个淋巴区域中,它的临床症状通常较轻、且与受影响的淋巴结有关,器官功能障碍少见。 UCD的病因学研究认为,UCD的发病可能与淋巴结对正常抗原刺激的过度反应有关,也可能与低级别的肿瘤形成有关,其具体发病机理还有待研究。 一项于2001-2009年在美国进行的流行病学调查显示,Castleman病年发病率约6500~7700人/年,其中约4900~5900例为UCD。UCD可以在任何年龄发生,但在年轻人中更常见,诊断中位年龄约为30~35岁。女性UCD的发病率略高于男性。 UCD患者通常没有明显的临床表现,大部分患者因在体格检查或影像学检查中发现淋巴结肿大而就诊。 在UCD诊断中,患者通常没有实验室指标异常,CT显示有中度及以上单个持续增大的淋巴结。确诊通常依靠肿大淋巴结的切除活检。 UCD的病理学特征为良性淋巴细胞的扩增,通常淋巴结的结构完整,B细胞和浆细胞为多克隆,T细胞没有异常免疫表型。 根据病理学表现UCD可分为三种亚型:透明血管型、浆细胞型和混合型。 UCD一般有极好的长期预后,诊断为UCD后,预期寿命通常不会改变。然而,研究显示UCD患者罹患副肿瘤性天疱疮和淋巴瘤的风险增高。手术切除肿大的淋巴结是目前UCD首选治疗方法,在无法切除的情况下通常采取局部放疗,95%的患者可以达到长期生存。 iMCD概述iMCD是Castleman病中一类病因不明且预后较差的亚型,其特征为累计多个淋巴区域的淋巴结肿大,临床表现多样,某些症状轻微,另一些则可能危及生命。 目前,iMCD的病因尚不清楚, 临床上多见与其他免疫学疾病重叠,这提示iMCD的发病可能涉及多个过程。目前研究认为iMCD发病机制可能与以下4个因素相关: 自身免疫机制:约30%的iMCD患者具有自身抗体和自身免疫性; 自身炎症机制: iMCD患者存在炎症反应相关的单基因突变; 肿瘤机制: iMCD具有与淋巴瘤重叠的临床和组织病理学特征,iMCD患者的恶性肿瘤发生率增加; 感染机制:iMCD与HHV-8相关MCD在临床和组织病理学上的表现相似,尽管研究未能在iMCD中未能检测到病毒RNA,但仍不能排除存在病毒感的间接作用。 流行病学调查显示,MCD患者约占CD患者23%,而iMCD患者约占MCD患者的33%~58%。在中国,iMCD/MCD的比例更高,达84%,以美国发病率数据估算中国每年iMCD新增病例为4600~5477。iMCD可发病于任何年龄段(2~80岁),确诊的中位年龄为50岁,11%患者发病年龄小于19岁,58%患者为男性。 iMCD临床表现多样,但也存在一些常见体征和实验室指标异常。几乎所有iMCD患者出现炎症性症状(包括盗汗、体重减轻、虚弱、疲劳、肝脾肿大、血细胞减少和器官功能障碍)和皮肤症状(包括皮疹、血管瘤和天疱疮)。 iMCD患者的实验室检查中通常表现出高炎症指标(高CRP、高ESR、高IL-6、高血小板、高纤维蛋白原和高IgG)和器官损害(贫血、低白蛋白血症、肾功能不全和高胆红素)的特点。 尽管存在一些常见症状和实验室检查结果, 不同的iMCD患者可以表现出非常不同的临床特征和实验室特征,同时,iMCD病理特征与多种疾病存在交叉,这导致了iMCD的诊断极其困难。调查显示,55% MCD患者曾被误诊,35%患者需经历1年甚至数年才能确诊。iMCD的诊断主要依赖于淋巴结活检,其次还包括实验室检查和临床症状,鉴别诊断需要排除感染相关性疾病、自身免疫性/自身炎症性疾病、恶性/淋巴增生性疾病等多种疾病。对于难以确诊的可疑患者可以考虑进行iMCD经验性治疗。 目前,用于治疗iMCD的药物包括IL-6抗体司妥昔单抗,化疗,糖皮质激素治疗和放疗。iMCD患者预后受分型和累及部位等多因素影响,但整体预后不良,美国这部分患者的5年死亡率为35%,中国为49%。据2020年美国血液学协会(ASH)年会上公布的一项真实世界数据显示,iMCD患者诊断后平均生存时间仅2.57年。 参考文献 1. Castleman B, Towne VW. Case records of the Massachusetts General Hospital: case no. 40231. N Engl J Med 1954;250:1001–5. 2. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020 Apr 16;135(16):1353-1364. 3. Fajgenbaum DC, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017 Mar 23;129(12):1646-1657. 4. Liu AY, et al. Idiopathic multicentric Castleman's disease: a systematic literature review. Lancet Haematol. 2016 Apr;3(4):e163-75. 5. Munshi N, et al. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015 May;56(5):1252-60. 6. Simpson D. Epidemiology of Castleman Disease. Hematol Oncol Clin North Am. 2018 Feb;32(1):1-10. 7. Van Rhee F, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018 Nov 15;132(20):2115-2124. 8. Zhang X, et al. Clinical characteristics and outcomes of Castleman disease: A multicenter study of 185 Chinese patients. Cancer Sci. 2018 Jan;109(1):199-206. 9. 贾鸣男,张路,李剑.特发性多中心型Castleman病的诊疗进展[J].中国肿瘤临床,2019,46(11):541-545. 10. 刘海玲,范磊,李建勇.Castleman病的诊疗进展[J].中华血液学杂志,2020,41(8):697-700.
 张路 副主任医师 北京协和医院 血液内科1.1万人已读
张路 副主任医师 北京协和医院 血液内科1.1万人已读 - 精选 特发性多中心型Castleman病的诊疗进展
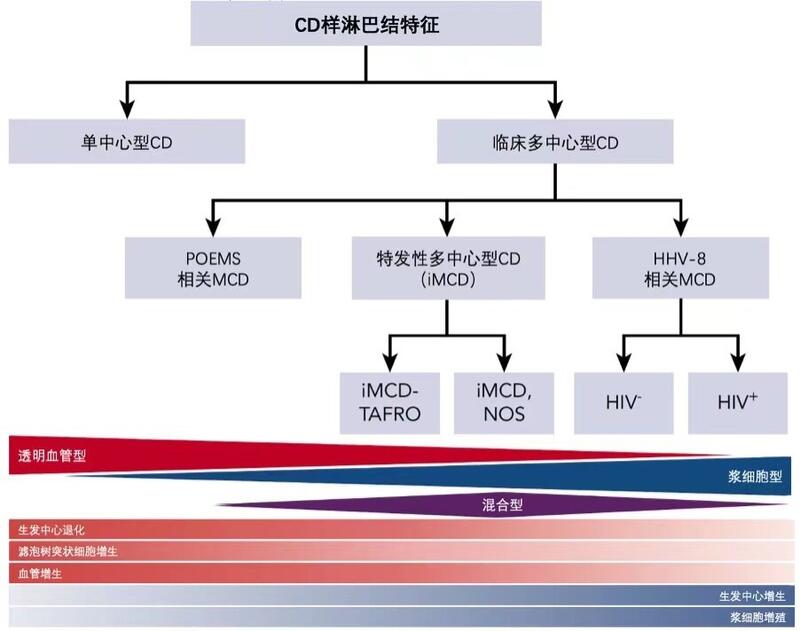
Castleman病(Castleman disease,CD)是一类具有特征性病理改变、高临床异质性的淋巴增生性疾病。2018年,该病被纳入国家卫健委《第一批罕见病目录》。根据淋巴结组织的病理学表现不同,可分为透明血管型、浆细胞型、混合型三型。根据淋巴结分布和器官受累部位的情况不同,又可分为单中心型Castleman病(unicentric Castleman disease,UCD)和多中心型Castleman病(multicentric Castleman disease,MCD)。前者发病率相对较高,仅累及单个淋巴结区域,全身症状反应较轻,主要依靠手术治疗,预后较好;后者发病率相对较低,累及多个淋巴结区域,多有全身症状、血细胞减少,甚至威胁生命的脏器功能受损,预后较UCD差[1]。2018年以前,MCD的治疗尚无“标准”方案[2],该病的诊疗存在较多问题。针对这些问题,2017年,国际上首次提出共识及诊断标准,根据是否感染人类疱疹病毒-8(human herpes virus-8,HHV-8)将MCD进一步分为HHV-8阳性的MCD——常见于人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染患者或免疫抑制人群[3,4],以及HHV-8阴性的MCD,也称为特发性MCD(idiopathic multicentric Castleman disease,iMCD)[5]。前者病因清楚,有明确有效的治疗手段[6],不是本综述关注的重点;后者占MCD患者中的33%-58%[7],且中国人中iMCD患者的比例比国外更高[8,9],可发生于任何年龄段,病因未明[10],预后较差(文献报道的5年生存率在55%-77%之间[11-13]),是近年来CD研究的热点和进展最迅速的领域。紧随2017年iMCD诊断标准的提出,2018年,针对iMCD缺乏治疗指南、诊疗较为混乱的现状,又提出了国际上首个iMCD的治疗共识,将该领域的诊疗又推至了一个新的高度[7]。尽管iMCD是近年来血液病中一个进展非常迅速的领域,但由于该病发病率低,国内医生对该病的认识尚有一定提升空间。本文旨在对iMCD领域近年来在诊断和治疗方面的进展进行综述和回顾,并介绍该领域的最新进展。1 iMCD的诊断进展iMCD的发病率低,临床表现多样,常与其他疾病重叠出现,缺少明确的生物标记物,临床医生对此病缺乏认识,使得iMCD的漏诊和误诊非常常见。基于文献复习和专家意见,2017年Castleman Disease Collaborative Network(CDCN)提出了统一的iMCD分类系统[5]:从淋巴结病理角度出发,根据临床表现将CD患者分为单中心型CD(UCD)或多中心型CD(MCD);根据病因,将MCD分为四类:①HHV-8相关的MCD,②POEMS综合征相关的MCD,③iMCD(病因未明),④其他具有类似CD淋巴结病理表现的疾病,即诊断iMCD时需要排除的感染、免疫、恶性肿瘤等疾病。iMCD的诊断主要依据于淋巴结病理、实验室标准及临床表现,2017年CDCN提出iMCD诊断标准的全球共识(见表1)[5]。关于此诊断标准,有以下几点要点需要注意:1.主要标准:淋巴结病理方面,透明血管型主要表现为生发中心退化(常伴有套区增生,“洋葱皮”样改变)和明显的滤泡树突状细胞增生;浆细胞型主要表现为生发中心增生及浆细胞增殖,混合型的组织学特征则介于以上两型之间;任何一种病理类型均可见血管插入生发中心[7]。另外,iMCD患者的组织病理标本的EB病毒编码RNA(Epstein-Barr encoded RNA, EBER)染色及潜伏期相关核抗原(latency-associated nuclear antigen,LANA-1)染色必须是阴性的。目前,病理类型与临床治疗之间的关系尚不明确。淋巴结受累个数方面,可行全身CT或18FDG-PET评估,后者也可以通过摄取水平与淋巴瘤相鉴别[14,15]。2.排除标准:很多疾病患者的淋巴结均可出现Castleman样的改变,这些疾病也称为“模拟”MCD的疾病。考虑到这些患者与iMCD患者可能具有不同的发病机制、临床表现,诊断iMCD时排除这些疾病则至关重要。详见表1“排除标准”。3.其他表现:血液中白细胞介素-6(interleukin-6,IL-6),可溶性白细胞介素-2受体(soluble interleukin-2 receptor,sIL-2R)、血管内皮生长因子(vascular endothelial growth factor,VEGF)、免疫球蛋白A(immunoglobulin A,IgA)、免疫球蛋白E(immunoglobulin E,IgE)、乳酸脱氢酶(lactate dehydrogenase,LDH)、β2微球蛋白(β-2-microglobulin,β2-MG)的水平升高、骨髓活检纤维化或多克隆浆细胞增多等表现对于iMCD的诊断也有一定的提示意义,但是由于证据不足,尚未纳入诊断标准中[5,16]。另外,由于IL-6的敏感性和特异性都不够强,IL-6水平未被纳入诊断标准[17]。根据患者临床特点不同,还可将iMCD进一步分为两种亚型:(1)TAFRO亚型:具有血小板减少(thrombocytopenia,T)、重度水肿(anasarca,A)、发热(fever,F)、骨髓纤维化(reticulin fibrosis of the bone marrow,R)和器官肿大(organomegaly,O)的特征,而丙种球蛋白水平正常,细胞因子多为VEGF升高而不是IL-6升高[18],淋巴结病理多为透明血管型或混合型。2017年,日本教授们提出了TAFRO综合征的诊断标准(见表2),此型患者临床表现更重,预后更差[16,19,20]。(2)非特指iMCD(iMCD-not otherwise specified,iMCD-NOS):患者炎症反应相对较轻,临床主要表现为IL-6升高、血小板正常或增多、多克隆免疫球蛋白升高,全身水肿少见,淋巴结病理为浆细胞型或混合型[5,20]。自2017年iMCD诊断标准发表后,目前已经得到国际血液学界的公认:一方面,纠正了该病临床研究入组标准不统一的问题,后续开展的该病临床研究均以该诊断标准作为入选患者的条件,为该罕见病临床研究的进一步深化提供了基础[21];另一方面,国际公认的诊断标准的建立,也给未来该病的规范化治疗奠定了基础。2 iMCD的治疗进展2.1 疾病严重程度评估对于明确诊断iMCD的患者,治疗前应进行疾病严重程度评估,从而更好地为患者选择治疗方案。根据2018年发表的治疗共识[7],可将重型患者定义为出现器官衰竭或需要重症监护的患者,可通过以下5条满足≥2条进行判断:①ECOG评分≥2分;②IV期肾功能不全(eGFR<30mL/(min·1.73m2)或血肌酐>3.0mg/dL);③全身水肿和/或腹水和/或胸膜/心包积液(高细胞因子血症/低白蛋白的影响);④血红蛋白≤8.0g/dL;⑤肺部受累/间质性肺炎伴呼吸困难。此类患者往往伴有器官功能衰竭的表现,且多为TAFRO亚型,病情危急。不符合以上标准的患者则称之为非重型患者。2.2 治疗药物2.2.1 IL-6靶向治疗:即抗IL-6的司妥昔单抗(Siltuximab)及抗IL-6受体的托珠单抗(Tocilizumab)[22,23]。文献报道,IL-6在iMCD患者的发病及临床表现中起着至关重要的作用[4];部分iMCD患者的症状严重程度与IL-6水平有一定的相关性;疾病活动时患者的IL-6水平也明显升高[24]。根据2018年的治疗共识,对于iMCD患者,无论病情严重与否,推荐使用司妥昔单抗或托珠单抗作为一线治疗[7]。司妥昔单抗是一种抗IL-6的人鼠嵌合免疫球蛋白G1κ(IgG1κ)型单抗。在iMCD患者中进行的唯一一项随机、双盲、安慰剂对照试验就是针对司妥昔单抗的。此项研究表明,使用司妥昔单抗(11mg/kg,每3周一次)的患者中,34%实现持续性肿瘤缓解及症状缓解,38%的患者实现了肿瘤缓解,约60%的患者实现持续的症状缓解;而安慰剂组的持续性肿瘤缓解及症状缓解率为0%,肿瘤缓解率为4%,持续性症状缓解率约为20%。使用司妥昔单抗的患者中,达到三级或以上的不良反应主要为疲劳及盗汗,少见严重的血液学或实验室检查异常,未发生严重的肝功能异常或胃肠道穿孔[23,25]。另外,司妥昔单抗治疗是否有效与基线IL-6水平无直接关系[24]。此药已在美国、加拿大、欧盟、巴西等多个国家批准用于治疗iMCD,遗憾的是,中国尚未引进此药。托珠单抗是一种人源化针对人IL-6受体的IgG1κ型单克隆抗体。2005年,Nishimoto和他的团队研究证明此药在MCD患者中的有效性及安全性,可改善患者的症状及多项实验室指标,且不良反应较轻[22]。Nishimoto团队后续报道,86%的患者对托珠单抗治疗持续有效,超过90%的患者IL-6水平升高,所有患者都有明显的全身炎症综合征[26]。因此,iMCD患者也可以选择托珠单抗(8mg/kg,每2周一次)进行治疗。由于托珠单抗用于iMCD治疗的相关文献较少,且缺乏随机对照研究,因此推荐等级(grade 2A)较司妥昔单抗(grade 1)低[7]。目前暂无文献比较司妥昔单抗和托珠单抗之间的疗效,可根据实际情况进行选择。2.2.2 糖皮质激素治疗:大剂量的糖皮质激素可抑制iMCD患者的高炎症、高细胞因子状态,但是由于糖皮质激素单药治疗的长期缓解率低、复发率高、治疗失败率高,因此并不推荐糖皮质激素单药治疗iMCD,可在治疗初期与其他药物联用以控制病情[12,27,28]。根据2018年治疗共识,对于非重型患者,治疗初期可根据患者病情决定是否加用糖皮质激素;而对于重型患者,可考虑大剂量糖皮质激素(如:甲泼尼龙500mg/天)与司妥昔单抗或托珠单抗联合使用作为一线治疗方案[7]。2.2.3 传统化疗(含或不含利妥昔单抗(Rituximab)):传统细胞毒性化疗方案虽然缓解率很高,但是复发率也很高,副作用明显,不推荐用于非重型患者;对于重型iMCD患者而言,糖皮质激素联合抗IL-6单抗治疗可能需要一定时间起效,部分患者甚至对此治疗无效,但此类患者随时有生命危险,因此,如果重型患者在司妥昔单抗治疗一周后无效或治疗未达一周出现病情恶化趋势时,应开始多药化疗以抑制免疫系统及细胞因子风暴,挽救患者的生命[7]。常用的化疗方案包括:R-CHOP(利妥昔单抗,环磷酰胺,多柔比星,长春新碱,泼尼松)、CVAD(环磷酰胺,长春新碱,阿霉素,地塞米松)、CVP(环磷酰胺、长春新碱、泼尼松)、VDT-ACE-R(硼替佐米、地塞米松、沙利度胺、多柔比星、环磷酰胺、依托泊苷、利妥昔单抗)、含依托泊苷/环磷酰胺的方案等[16,27,29]。而针对HHV-8阴性MCD,即iMCD患者的研究中,利妥昔单抗多与传统化疗药物联合使用。曾有文献统计,使用利妥昔单抗或以利妥昔单抗为基础的化疗作为一线治疗的iMCD患者,完全缓解率和部分缓解率分别为20%和48%,但使用利妥昔单抗的无进展生存期较司妥昔单抗短[21]。另一部文献中统计,8位使用利妥昔单抗单药治疗的患者中,有5位患者达到完全缓解,只有1位治疗失败[28]。在国内一项回顾性研究中,27名患者进行了至少两个疗程以利妥昔单抗为基础的化疗方案治疗,完全缓解率和部分缓解率分别为33%和22%,难治-复发型患者(两个疗程化疗后未达到部分缓解,或完全缓解/部分缓解后3个月后复发)的完全缓解率和部分缓解率分别为14%和28%[9]。因此,结合2018年的治疗共识,对于非重型患者,可以考虑使用利妥昔单抗(375mg/m2×4-8个疗程)作为IL-6靶向治疗的一线替代方案,或联合激素和/或免疫调节剂作为IL-6靶向治疗失败后的二线方案[7]。2.2.4 免疫调节治疗:包括沙利度胺、环孢素A、西罗莫司、来那度胺、硼替佐米、白细胞介素-1受体拮抗剂(阿那白滞素)、维甲酸衍生物、干扰素-α,均有个案报道治疗iMCD患者有效。沙利度胺作为一种免疫调节剂,可以抑制白细胞介素-1、IL-6、VEGF、肿瘤坏死因子-α等多种细胞因子的产生,在个案报道中沙利度胺对iMCD患者治疗有效[30-32]。北京协和医院近期报道的一项二期临床试验,在iMCD患者中使用口服TCP方案(沙利度胺100mg/日×2年,环磷酰胺300mg/m2/周×1年,泼尼松1mg/kg每周两次×1年)治疗,48%的患者实现了24周及以上的症状缓解及肿瘤缓解,1年总生存期及1年无进展生存率分别为88%和60%(司妥昔单抗的随机对照试验中1年总生存期及1年无进展生存率分别为100%和60%);副反应方面,肺部感染致死一例,三级及以上副反应出现皮疹一例,总体来讲治疗安全有效,为无法获得IL-6靶向治疗的初治患者提供了治疗的新思路[33,34]。另外,对于非重型患者,2018年的治疗指南推荐在IL-6靶向治疗及利妥昔单抗治疗均失败后,也可以使用免疫调节药物进行治疗[7]。2.3 TAFRO综合征的治疗在日本的一个25例TAFRO综合征患者的队列中,23/25例患者一线治疗选择了糖皮质激素,其中11/23(47.8%)的患者对糖皮质激素治疗反应良好,难治性患者可能需要联合其他治疗,3例患者死亡(2例疾病进展和1例败血症)[16]。也有其他专家提出将足量糖皮质激素作为TAFRO综合征患者的一线治疗,病情危急的患者可选择激素冲击治疗[35]。根据2018年的治疗共识,建议TAFRO综合征患者同其他iMCD患者一样,先进行疾病严重度评估,进而决定治疗方案;一线治疗方案仍可选择IL-6靶向治疗联合或不联合糖皮质激素[7]。根据既往研究,环孢素A可作为TAFRO综合征患者在IL-6靶向治疗失败后的二线方案,有助于改善难治性腹水及血小板减少的情况[21,36-38]。由于TAFRO综合征患者中IL-6升高水平不突出[22],蛋白质谱与其他iMCD患者也不同[39],更多的发病机制有待于我们探索。3 结论与展望iMCD是一类异质性很强、预后较差的罕见病。最近几年,iMCD领域进展迅速,有了全新的诊断标准和治疗共识,为后续的病因研究及临床研究提供了坚实的基础。对中国的临床医生而言,在结合国际专家意见的基础上,可根据实际情况为患者选择经济、有效的治疗方案。表1 iMCD的诊断标准主要标准(满足2条)1.淋巴结组织病理学符合iMCD改变 2.至少两处淋巴结肿大(短轴≥1cm)次要标准(11条中至少满足2条,且至少1条为实验室标准)实验室标准: 1.C反应蛋白升高或血沉增快* 2.贫血 3.血小板减少或增多 4.低白蛋白血症 5.肾功能不全或蛋白尿 6.多克隆免疫球蛋白升高临床标准: 1.盗汗、发热、体重下降、乏力等全身症状 2.肝肿大和/或脾肿大 3.浆膜腔积液或全身水肿 4.皮疹(樱桃样血管瘤或紫罗兰皮疹) 5.淋巴细胞间质性肺炎排除标准(以下疾病均需排除) 感染相关:HHV-8、EBV、CMV、弓形虫、HIV、活动性结核 自身免疫病/自身炎症性疾病(需满足诊断标准,仅有自身免疫抗体阳性不能排除诊断):系统性红斑狼疮,类风湿性关节炎,成人Still病,幼年特发性关节炎,自身免疫性淋巴增生性综合征 恶性肿瘤/淋巴增生性疾病:淋巴瘤(霍奇金淋巴瘤,非霍奇金淋巴瘤)、多发性骨髓瘤、原发性淋巴结浆细胞瘤、滤泡树突状细胞肉瘤、POEMS综合征CMV,巨细胞病毒;EBV,Epstein-Barr病毒;HHV-8,人类疱疹病毒-8;HIV,人类免疫缺陷病毒。*首选C反应蛋白作为疾病评估,无法测C反应蛋白时可用血沉替代。引自Fajgenbaum D C et al[5]。表2 TAFRO综合征的诊断标准组织病理学(满足以下两条) 1.淋巴结组织病理学符合TAFRO综合征表现 2.HHV-8相关的LANA-1染色阴性主要标准(满足以下三条) 1.满足TAFRO症状中至少3条:血小板减少(T),重度水肿(A),发热(F),骨髓纤维化(R)和器官肿大(O)* 2.无高种丙球蛋白血症 3.病变淋巴结体积较小次要标准(至少满足一条) 1.骨髓中巨核细胞偏高/正常 2.血清ALP升高且不伴血清转氨酶明显升高排除标准:排除感染、自身免疫/自身炎症性疾病、恶性肿瘤等疾病(见表1“排除标准”)ALP,碱性磷酸酶;HHV-8,人类疱疹病毒-8;LANA,潜伏期相关核抗原。*血小板减少定义为血小板计数<100×109/L,重度水肿定义为CT可见的胸腔积液和腹水,发热指体温>38℃,器官肿大指淋巴结或肝或脾肿大引自Iwaki N et al[16]。(原文刊载于《中国肿瘤临床杂志》,作者贾鸣男、张路、李剑)参考文献[1] Zhang L, Li Z, Cao X, et al. Clinical spectrum and survival analysis of 145 cases of HIV-negative Castleman's disease: renal function is an important prognostic factor[J]. Sci Rep, 2016, 6: 23831.[2] 张路, 李剑. 多中心型Castleman病的治疗进展[J]. 国际药学研究杂志, 2017, 44(2): 162-165.[3] Fajgenbaum D C, Van Rhee F, Nabel C S. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy[J]. Blood, 2014, DOI: 10.1182/blood-2013-12-545087.[4] Van Rhee F, Greenway A, Stone K. Treatment of Idiopathic Castleman Disease[J]. Hematol Oncol Clin North Am, 2018, 32(1): 89-106.[5] Fajgenbaum D C, Uldrick T S, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease[J]. Blood, 2017, DOI: 10.1182/blood-2016-10-746933.[6] Uldrick T S, Polizzotto M N, Aleman K, et al. Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease[J]. Blood, 2014, DOI: 10.1182/blood-2014-07-586800.[7] Van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease[J]. Blood, 2018, DOI: 10.1182/blood-2018-07-862334.[8] Dong Y, Wang M, Nong L, et al. Clinical and laboratory characterization of 114 cases of Castleman disease patients from a single centre: paraneoplastic pemphigus is an unfavourable prognostic factor[J]. Br J Haematol, 2015, DOI: 10.1111/bjh.13378.[9] Dong Y, Zhang L, Nong L, et al. Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease[J]. Ann Hematol, 2018, DOI: 10.1007/s00277-018-3347-0.[10] 张路, 李剑. Castleman病发病机制研究进展[J]. 中国医学科学院学报, 2016, 38(1): 118-121.[11] Dispenzieri A, Armitage J O, Loe M J, et al. The clinical spectrum of Castleman's disease[J]. Am J Hematol, 2012, DOI: 10.1002/ajh.23291.[12] Seo S, Yoo C, Yoon D H, et al. Clinical features and outcomes in patients with human immunodeficiency virus-negative, multicentric Castleman's disease: a single medical center experience[J]. Blood Res, 2014, 49(4): 253-258.[13] Melikyan A L, Egorova E K, Kovrigina Capital A C E M C, et al. [Clinical and morphological features of different types of Castleman's disease][J]. Ter Arkh, 2015, 87(7): 64-71.[14] Lee E S, Paeng J C, Park C M, et al. Metabolic characteristics of Castleman disease on 18F-FDG PET in relation to clinical implication[J]. Clin Nucl Med, 2013, 38(5): 339-342.[15] Kligerman S J, Auerbach A, Franks T J, et al. Castleman Disease of the Thorax: Clinical, Radiologic, and Pathologic Correlation: From the Radiologic Pathology Archives[J]. Radiographics, 2016, 36(5): 1309-1332.[16] Iwaki N, Fajgenbaum D C, Nabel C S, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease[J]. Am J Hematol, 2016, 91(2): 220-226.[17] Nishimoto N, Terao K, Mima T, et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease[J]. Blood, 2008, DOI: 10.1182/blood-2008-05-155846.[18] 张路, 李剑, 冯俊, et al. TAFRO综合征一例报告[J]. 中国医学科学院学报, 2016, 38(4): 484-486.[19] Iwaki N, Sato Y, Takata K, et al. Atypical hyaline vascular-type castleman's disease with thrombocytopenia, anasarca, fever, and systemic lymphadenopathy[J]. J Clin Exp Hematop, 2013, 53(1): 87-93.[20] Igawa T, Sato Y. TAFRO Syndrome[J]. Hematol Oncol Clin North Am, 2018, 32(1): 107-118.[21] Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease[J]. Blood, 2017, DOI: 10.1182/blood-2016-11-748855.[22] Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease[J]. Blood, 2005, DOI: 10.1182/blood-2004-12-4602.[23] Van Rhee F, Wong R S, Munshi N, et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial[J]. Lancet Oncol, 2014, DOI: 10.1016/S1470-2045(14)70319-5.[24] Casper C, Chaturvedi S, Munshi N, et al. Analysis of Inflammatory and Anemia-Related Biomarkers in a Randomized, Double-Blind, Placebo-Controlled Study of Siltuximab (Anti-IL6 Monoclonal Antibody) in Patients With Multicentric Castleman Disease[J]. Clin Cancer Res, 2015, DOI: 10.1158/1078-0432.CCR-15-0134.[25] Van Rhee F, Casper C, Voorhees P M, et al. A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease[J]. Oncotarget, 2015, 6(30): 30408-30419.[26] Matsuyama M, Suzuki T, Tsuboi H, et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman's disease[J]. Intern Med, 2007, 46(11): 771-774.[27] Herrada J, Cabanillas F, Rice L, et al. The clinical behavior of localized and multicentric Castleman disease[J]. Ann Intern Med, 1998, 128(8): 657-662.[28] Liu A Y, Nabel C S, Finkelman B S, et al. Idiopathic multicentric Castleman's disease: a systematic literature review[J]. Lancet Haematol, 2016, DOI: 10.1016/S2352-3026(16)00006-5.[29] Zhang L, Jiao L, Wang S J. Successful Treatment with Rituximab in a Patient with Castleman's Disease Complicated by Systemic Lupus Erythematosus and Severe Autoimmune Thrombocytopenia[J]. Chin Med J (Engl), 2015, 128(18): 2551-2552.[30] Lee F C, Merchant S H. Alleviation of systemic manifestations of multicentric Castleman's disease by thalidomide[J]. Am J Hematol, 2003, DOI: 10.1002/ajh.10310.[31] Tatekawa S, Umemura K, Fukuyama R, et al. Thalidomide for tocilizumab-resistant ascites with TAFRO syndrome[J]. Clin Case Rep, 2015, DOI: 10.1002/ccr3.284.[32] Ma W L, Zhang L, Zhu T N, et al. TAFRO Syndrome - A Specific Subtype of Castleman's Disease in China[J]. Chin Med J (Engl), 2018, 131(15): 1868-1870.[33] Zhang L, Zhao A L, Duan M H, et al. Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease[J]. Blood, 2019, DOI: 10.1182/blood-2018-11-884577.[34] Van Rhee F, Stone K. Storming the Castle with TCP[J]. Blood, 2019, 133(16): 1697-1698.[35] Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version[J]. Int J Hematol, 2016, DOI: 10.1007/s12185-016-1979-1.[36] Konishi Y, Takahashi S, Nishi K, et al. Successful Treatment of TAFRO Syndrome, a Variant of Multicentric Castleman's Disease, with Cyclosporine A: Possible Pathogenetic Contribution of Interleukin-2[J]. Tohoku J Exp Med, 2015, 236(4): 289-295.[37] Yamaga Y, Tokuyama K, Kato T, et al. Successful Treatment with Cyclosporin A in Tocilizumab-resistant TAFRO Syndrome[J]. Intern Med, 2016, 55(2): 185-190.[38] Takasawa N, Sekiguchi Y, Takahashi T, et al. A case of TAFRO syndrome, a variant of multicentric Castleman's disease, successfully treated with corticosteroid and cyclosporine A[J]. Mod Rheumatol, 2019, DOI: 10.1080/14397595.2016.1206243.[39] Iwaki N, Gion Y, Kondo E, et al. Elevated serum interferon gamma-induced protein 10 kDa is associated with TAFRO syndrome[J]. Sci Rep, 2017, 7: 42316.
张路 副主任医师 北京协和医院 血液内科28人已购买 - 精选 卡斯尔门病(Castleman disease)简介
卡斯尔门病(Castleman disease,CD)北京协和医院 李剑 张路 吴琪1、CD是什么?卡斯尔门病(又称血管滤泡性淋巴结增生)是由美国病理学家本杰明.卡斯尔门(Benjamin Castl
张路 副主任医师 北京协和医院 血液内科268人已购买 - 精选 帮您看懂体检报告中的血常规结果
目前很多人都会常规体检,而血常规化验单几乎是任何一个体检机构都会进行的检查,这个检查虽然便宜,但指标却非常多,往往有箭头,让人好不担心。那么,今天我们就来简单说说怎么来看血常规检查。1. 什么是血常规检查?北京协和医院血液内科张路血常规检查指通过观察血细胞的数量变化及形态分布从而判断血液状况及疾病的检查。是最“简单”、最便宜、最便捷的抽血检查,在医疗和体检服务中均广泛使用,常作为疾病诊断的一种辅助手段。2. 是不是必须每项都在正常范围内,我才算是“正常”?不是。一个血常规化验通常不到20块钱,里面却蕴藏20多个项目,下面这张化验单(图1),数数共28个项目,每个项目都有相应的正常值,若您不是处女座,真心不必强求每项都在正常范围内。血常规检查的项目非常多,大部分指标的所谓“正常值”是根据健康人群的一般分布而言的。可以用人的身高做个类比:如果把人群的身高分布来设定一个“正常值”,虽然在身高高于正常值的人里会存在罹患“巨人症”的个体,但也会存在诸如姚明这样的健康人。同理,血常规检查项目也是如此,很多时候,轻度的偏移所谓“正常值”是没有临床意义的。但显著的偏移(例如图1中的白细胞就是正常值的接近5倍),就像出现身高10米的人一样,往往是存在问题的。3.白细胞高=炎症吗?不是。虽然有时细菌感染(俗称的“有炎症”)会引起白细胞升高,但白细胞升高并不一定都是“炎症”(细菌感染)。这需要分成三个层面:首先,血常规里的“白细胞”是“淋巴细胞”,“单核细胞”,“中性粒细胞”,“嗜酸性粒细胞”,“嗜碱性粒细胞”这五个成分的总和,而这些细胞占白细胞的比例则是化验单中相应的“百分比”(图1),因此,这五个成分中任何一个成分的升高,都可能会引起白细胞总数的升高。大多数情况下,“淋巴细胞”、“单核细胞”、“嗜酸性粒细胞”、“嗜碱性粒细胞”升高都跟“炎症”没有太大关系。以图1为例,您可以看到白细胞明显升高 47.24(正常范围3.50-9.50),但细看后发现,最主要升高的成分是“淋巴细胞”(绝对值37.25,正常范围0.8-4.0)。事实上,这个血常规是慢性淋巴细胞白血病患者的结果,白细胞高是肿瘤细胞增殖的表现,并非“炎症”;其次,即使是细菌感染,也不都是白细胞高,有时候细菌感染反倒会引起白细胞降低;第三,虽然细菌感染常常会引起“中性粒细胞”升高,从而引起白细胞升高,但其它疾病(非“炎症”),例如骨髓增殖性肿瘤,也会引起中性粒细胞升高。综上可见,白细胞高并不一定是炎症,千万不要一看到白细胞高就用“消炎药”。4.单靠血常规,不借助其它检查,就能诊断某些疾病吗?也对也不对。血常规虽然简单、便捷,但蕴藏着非常丰富的信息量,尤其是与患者的病史相结合,有经验的大夫经常可以较为准确的进行诊断。下面就举几个例子:这是一张6个月女婴的血常规化验单,生长发育正常,体检发现轻度贫血(血红蛋白111g/L),红细胞平均体积(MCV)明显降低52.3fl。祖籍广西,母亲血常规有类似表现。这时,有经验的医生就基本可以为患者诊断“轻型地中海贫血”了。这是一张26岁女性的血常规化验单,近期素食减肥,平时月经量大,血常规发现贫血(血红蛋白73g/L),平均红细胞体积减小(66.1fl)。两年前体检无贫血。根据病史和血常规检查结果,即使不再做其它检查,诊断“缺铁性贫血”也没有太大问题了。当然,虽然诸如上面的情形,有经验的医生就可以单凭一张血常规做出相对准确的诊断,但考虑到疾病的复杂性,很多情况下单凭一张血常规很难做出正确的诊断。例如淋巴细胞升高,背后可能是病毒感染,也可能是慢性淋巴细胞白血病,也可能是淋巴瘤白血病。这时,就需要依靠血常规的提示,再进一步挑选合适的其它检查项目来帮助诊断。★张路医生温馨提醒★这篇小文号称帮您“看懂”血常规,但其实血常规蕴藏着非常丰富的信息,不是三言两语能够说清,甚至也不是每个医生都能“看懂”貌似简单的血常规检查。因此,如果您发现血常规有异常,还是建议去血液科门诊咨询,相信血液科大夫会给您专业和耐心的解答。
张路 副主任医师 北京协和医院 血液内科58人已购买 - 引用 血液科张路大夫:我对Castleman病的未来展望
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科974人已读 - 引用 血液科张路大夫:Castleman病的国际交流
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科751人已读 - 引用 血液科张路大夫:我与Castleman病的患者故事
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科818人已读 - 引用 血液科张路大夫:Castleman病治疗的解决方案
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科951人已读 - 引用 血液科张路大夫:Castleman病治疗的经验与问题总结
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科755人已读 - 引用 血液科张路大夫:真正与Castleman病结缘
卡斯特曼病系列专访
张路 副主任医师 北京协和医院 血液内科704人已读

张路副主任医师
北京协和医院血液内科

